This website uses cookies to ensure you get the best experience on our website.
- Table of Contents

Facts about Neurofibromin.
Stimulates the GTPase activity of Ras.
NF1 shows increased affinity for Ras GAP, but lower specific activity.Might be a regulator of Ras activity. .
| Human | |
|---|---|
| Gene Name: | NF1 |
| Uniprot: | P21359 |
| Entrez: | 4763 |

| Belongs to: |
|---|
| No superfamily |

DKFZp686J1293; FLJ21220; Neurofibromatosis-related protein NF-1; neurofibromin 1; neurofibromin; NFNS; VRNF; WSS
Mass (kDA):
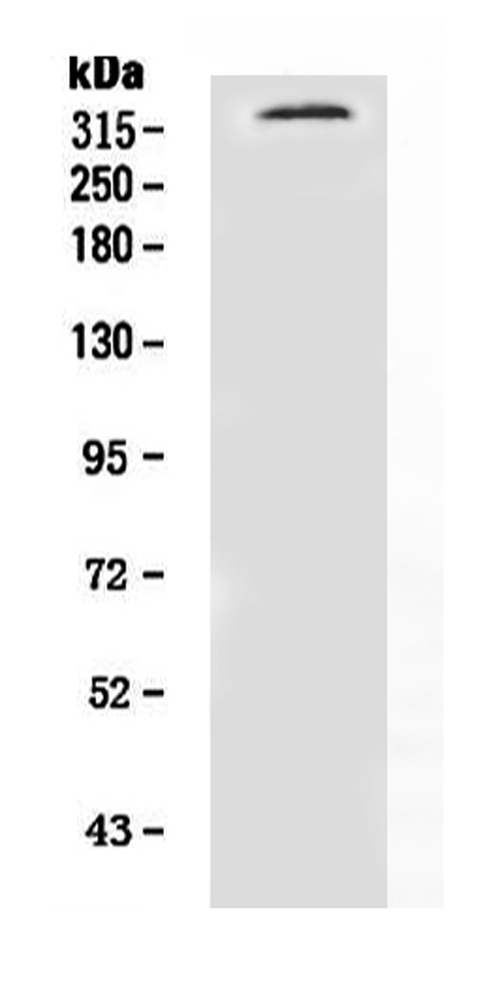
319.372 kDA

| Human | |
|---|---|
| Location: | 17q11.2 |
| Sequence: | 17; NC_000017.11 (31094927..31377677) |
Detected in brain, peripheral nerve, lung, colon and muscle.
Nucleus. Nucleus, nucleolus.



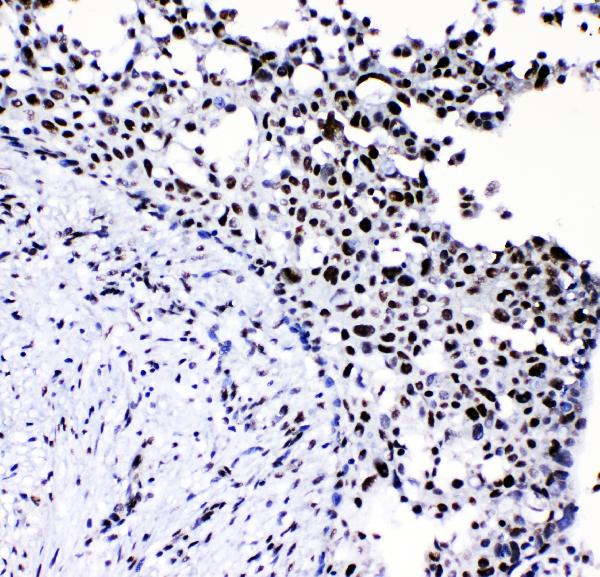
The NF1 Marker is genetically engineered, a gene associated with benign peripheral nerve-sheath tumors. Its high-affinity and high specificity make it a useful marker for the investigation of astroglia in the brain. For this purpose, Boster has validated all of its antibodies by testing known positive and negative samples to ensure high-specificity and high-affinity. This helps scientists apply product credits to any product that they use.
The mutation of NF1 is an uncommon, but potentially fatal, genetic disorder. It affects a single gene, and is usually hereditary. People with the disorder have a 50 percent chance of passing on their abnormal copy to their children. Moreover, this mutation has been associated with a range of other health issues, including learning disabilities and dysmorphisms. However, doctors are uncertain about the cause of the mutation.
The role of NF1 in malignancy is unknown, but it has been linked to a higher risk of neurofibromas. In fact, an epidemiologic study of 8,003 NF1 patients found an increased risk of various cancers. Of this group, 4% develop cancer. The incidence of chronic myeloid leukemia (CML) and female breast cancer (FBC) is 2.3% and 2%, respectively.
The NF1 mutation has a biallelic loss in melanocytes. Children with NF1 have a rare prevalence of CALMs, but CALMs are the most common pigmentary phenotype in patients with NF1 disease. Diagnoses for NF1 in young children are often made after clinical signs and symptoms are present. Genetic counseling is essential to discuss the risk of severe phenotypes and malignancy in a patient with NF1 mutation.
While various murine models have been developed to study NF1 mutations, none of them recapitulates the full spectrum of disease seen in NF1 patients. The traditional germ line heterozygous knockout mouse did not develop neurofibromas and tumors associated with NF1, but a subset of mice developed less common tumors later in life. Mice models have been developed that replicate more common NF1 features. Cre-lox mouse models have been developed to produce mice that have bi-allelic loss of Nf1 in a specific cell lineage. In addition, double mutant Nf1/Tp53 mice were developed to study tumors associated with malignant peripheral nerve sheath.
In a recent study, researchers isolated a recurrent nonsense mutation within the NF1 gene. This mutation is located in exon 41 of the swine NF1 gene and shares 100% amino acid identity with human exon 39. TALENs flanking the mutation were transfected into fetal Ossabaw minipig fibroblasts, and colonies were genotyped for NF1R1947*.
Although NF1 has several effects on human health, its expression is very variable and has even been linked to siblings. The clinical phenotype in siblings with NF1 should be the same in affected siblings. Furthermore, the disease is not caused by mutations in a single gene. Instead, other loci may be involved, as suggested by this study. So, scientists will continue to study NF1 in more detail.
NF1 mutations can result in optic pathway gliomas. Approximately 15% of young children with NF1 mutations develop these tumors. The majority of patients with this condition are under seven years old. In addition, the symptoms of OPG include unilateral proptosis, poor visual acuity, and field defects. The risk of developing optic pathway gliomas (OPG) is unpredictable, and frequent monitoring is necessary for a cure.
The gene NF1 is associated with malignant peripheral nerve-sheath tumors. The researchers used cyclooxygenase-2 inhibitors to induce apoptosis in human malignant peripheral nerve-sheath tumor cells. The study was published in PLOS ONE. The authors used sulindac derivatives to inhibit the growth of these tumor cells and induce apoptosis.
NF1 is the cause of plexiform neurofibromas in 50% of patients. These tumors arise from peripheral nerves and involve multiple fascicles. These tumors grow along the nerves and may collide, preventing complete surgical resection. This tumor is often associated with pain. In the majority of cases, the tumor is non-metastatic. However, treatment options for NF1 tumors vary.
The most common malignancy in NF1 patients is MPNST. This is the only type of peripheral nervous system cancer that is associated with NF1. Although other types of cancers have the same location, the nerves of the peripheral nervous system are involved in a variety of processes, including metastasis, lymphomas, and tumors of the central nervous system. Most MPNSTs consist of spindle cells and are characterized by their cellularity and morphology. Low-grade MPNST may contain mitotic figures and necrosis. Because this tumor typically develops in peripheral nerves, considerable tissue heterogeneity complicates diagnosis.
Inactivation of PRC2 boosts the Ras signaling pathway. This pathway is already activated in MPNST. The decreased levels of this gene explain the low rates of MPNST in patients with NF1.
Although most cases of peripheral nerve-sheath tumors are benign, the exact cause is not known. Some of them are hereditary and may occur in people with certain family members. The most common type is schwannoma, which forms inside the nerve. Symptoms of peripheral nerve-sheath tumors include pain, weakness, and loss of muscle control.
There are a number of other genetic mutations associated with NF1 in MPNST. Those responsible for NF1-derived MPNST have a low-expression of certain miRNAs. Moreover, a small number of paired samples may make it impossible to establish any consistent up or downregulation of specific miRNAs. The study highlights the importance of paired samples and well-characterized cohorts.
The high internal tumor burden in patients with NF1 is associated with an increased risk for MPNST. Furthermore, MRI has been used to identify subgroups with high internal tumor burden. Previous studies have also found a significant association between high internal tumor burden and increased risk for MPNST. Additionally, patients with higher internal tumor burden were found to have a significantly higher whole-body tumor volume.
The research team at MD Anderson in Houston, Texas, studied 1,600 patients with NF1 between 1985 and 2020. They compared the cancer survival rate of these individuals to that of the general population. They found that those with NF1 had a 10 times higher risk for cancer than the general population. It's unclear whether the association is true in all patients, but the research highlights the importance of knowing the genetic code of a patient's cancer.
NF1 is expressed by neurofibroblasts derived from tumors. The researchers used actin-normalized optical density units (ANUs) to calculate NF1 expression levels. The control Schwann cells were derived from normal nerves and NF1 mRNA was absent in them. In contrast, tumor-derived fibroblasts were enriched in NF1 mRNA.
Although the study found a significant association between NF1 microdeletions and the tumor burden of a patient with a specific NF1 mutation, there are many limitations in these studies. Because NF1 microdeletions can affect other genetic markers, these studies are best performed on patients with NF1 mutations, although further research is required. And, a single copy of the gene encoding the NF1 protein has a high frequency of copy number variations.
Mutations in NF1 can result in chromosome instability and aneuploidy. Thus, the genetic code of a tumor may be altered by homologous recombination or cumulative damage. For example, tumors with large constitutional deletions may have an increased risk of NF1 mutations. However, the same deletions may not be present in a single patient. If this is the case, the gene will be affected by several other genetic factors as well, such as tumor invasion.
NF1 inactivation is more common in S100+ cells compared to fibroblasts. This suggests that cell-specific differences may determine NF1 susceptibility. For example, S100+ cells may be monoclonal, whereas those of polyclonal nature may be due to nonrandom mutations. To gain a better understanding of neurofibroma formation, we must determine the status of both NF1 alleles in tumor-derived cell populations.
The S100+ cells in neurofibromas are widely assumed to be Schwann cells. In two cases, they were confirmed by co-expression of myelin P0. Interestingly, NF1 expression levels in different neural crest derivatives may depend on developmental timing. Inhibition of NF1 may result in heart defects in patients with NF1 microdeletion. Further research is needed to determine if NF1 deficiency is a causal factor in heart defects.
Microscopic NF1 microdeletions are rare and occur in only one-fourth of patients with gross NF1-repeats. They are mediated by NAHR between NF1-REPb and NF1-REPc. Interestingly, these patients exhibit hemizygosity in nine protein-coding genes. Atypical large NF1 deletions are quite heterogeneous and contain several genes.
PMID: 1457041 by Bernards A., et al. Complete human NF1 cDNA sequence: two alternatively spliced mRNAs and absence of expression in a neuroblastoma line.
PMID: 2134734 by Wallace M.R., et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients.