This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Let Bosterbio handle your Western blot CRO services and be your long term partner in broadening the bandwidth of your lab/team in a flexible manner.
The first step of gel preparation is to determine the gel percentage based on the molecular weight of your protein sample:
| Protein Size (kDa) | >100 | 30-100 | 10-30 | <10 |
|---|---|---|---|---|
| Gel Percentage | 8% | 10% | 12% | 15% |
If you are not sure of the size of your protein or are looking at proteins of a variety of molecular weights, then a gradient gel may provide the best resolution.
Notes
Dual Color Protein Loading Buffer is designed to prevent protein degradation during sample heating prior to electrophoresis and is able to work against pH changes during SDS-PAGE run. Many proteins are sensitive to pH changes that result from temperature fluctuations of Tris buffers during electrophoresis. It contains two tracking dyes: blue (Bromophenol Blue) for tracking the progress of electrophoresis and pink (Pyronin Y) for monitoring protein transfer to the membrane. Refer to the datasheet on our website for more information.
After electrophoresis, we recommend using one of our gel staining solutions to determine if the electrophoretic separation worked. Please refer to the datasheet(s) on our website for more information.
Note: Stained gel cannot be used in the subsequent protein transfer procedure.

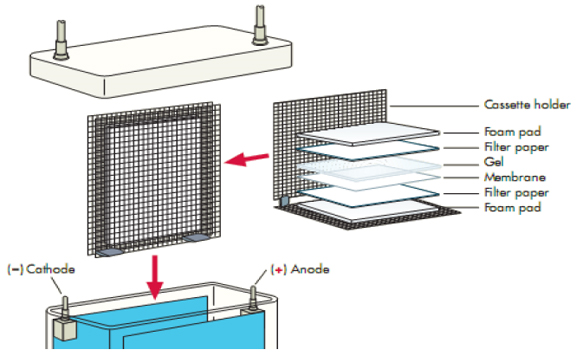
We recommend using one of the filter papers from Boster:
After blocking, the membrane is incubated with a primary antibody (that binds to the target protein) followed by an enzyme conjugated secondary antibody (HRP- or AP-conjugated secondary antibody).
The choice of western blot antibody detection methods depends on specific substances that are labeled or bound to the secondary antibody. ECL chemiluminiscent detection system and DAB chromogenic detection system are typically used in western blotting.
In this section, we provide the protocols for the Enhanced Chemiluminescence Detection (ECL) and colorimetric detection (DAB) methods. Use the method that fits your preferences and criteria.
Boster ECL chemiluminescent system depends on incubation of the western blot with a substrate that will luminesce when exposed to HRP on the secondary antibody. The light is then detected and captured by photographic film.
| Origin of Primary Antibody Species | Catalog # of ECL Kit* |
|---|---|
| Mouse IgG | EK1001 |
| Rabbit IgG | EK1002 |
| Goat IgG | EK1003 |
| Rat IgG | EK1004 |
| Mouse IgM | EK1005 |
* Each kit has sufficient reagents for 800 cm2 of membrane.
† Instead of using the ECL kit which provides 1) chromogenic reagents A and B (20X concentrated; 5 mL), 2) blocking buffer and 3) HRP-conjugated secondary antibody, one may use one of the following standalone chromogenic reagents A and B from Boster:
| Reagent A | Reagent B | ||||
|---|---|---|---|---|---|
| Product | Conc. | Volume | Conc. | Volume | Catalog # |
| ECL Western Blotting Substrate | 1X** | 100 mL | 1X** | 100 mL | AR1170 |
| Hypersensitive WB Chemiluminescent Substrate | 20X | 5 mL | 20X | 5 mL | CR0001-5 |
| Hypersensitive WB Chemiluminescent Substrate | 20X | 10 mL | 20X | 10 mL | CR0001-10 |
| Hypersensitive WB Chemiluminescent Substrate | 20X | 25 mL | 20X | 25 mL | CR0001-25 |
** Ready-to-use
Record the test result using autoradiography film or CHEMIDOC in a darkroom. For autoradiography film, exposure time for the specific antigen depends on the developing effect, which could range from seconds to minutes.
Using WB Developing Fixing Kit (Boster Catalog # AR0132), develop and fix the film in a dark room immediately. Alternatively, fluorescence CCD scan, digital imager, or luminometer can be used.
Notes
Prepare DAB or BCIP/NBT substrate solution described below.
| Origin of Primary Antibody Species | Color | Catalog # of DAB Kit |
|---|---|---|
| Mouse IgG | Yellow | SA2020 |
| Goat IgG | Yellow | SA2021 |
| Rabbit IgG | Yellow | SA2022 |
| Rat IgG | Yellow | SA2023 |
| Mouse IgG | Blue | SA2024 |
| Rabbit IgG | Blue | SA2025 |
Unleash the power of our exceptional service to accelerate your research and unlock valuable insights from your samples. Reach out to us now to discuss your project requirements and witness the transformative impact our our expert Western blotting service.
Learn the concept behind Western blotting. It is a technique that is used to detect specific proteins in the given sample. It usually involves two major processes, namely, SDS-polyacrylamide gel electrophoresis and protein blotting and testing.
Learn ELISA PrincipleCheck out this Western Blot sample preparation guides to learn how to get the best results from your sample type. Learn more about sample preparation in this guide.
Learn Western Blot Sample PreparationGet to learn Pro tips on resolving common Western Blot issues such as weak signal, wrong band size, smiley gel, and high background.
Learn Western Blot Troubleshooting TipsGet to learn the concept behind our best practises on Western Blot optimization. Learn how to optimize every aspect of your experiment to yield the best results.
Browse Western Blot Optimization Tips