This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
Alzheimer’s disease (AD) is characterized by an irreversible decline in cognitive and behavioral abilities that will eventually render the person completely reliant on others for basic activities in daily life.
View pathway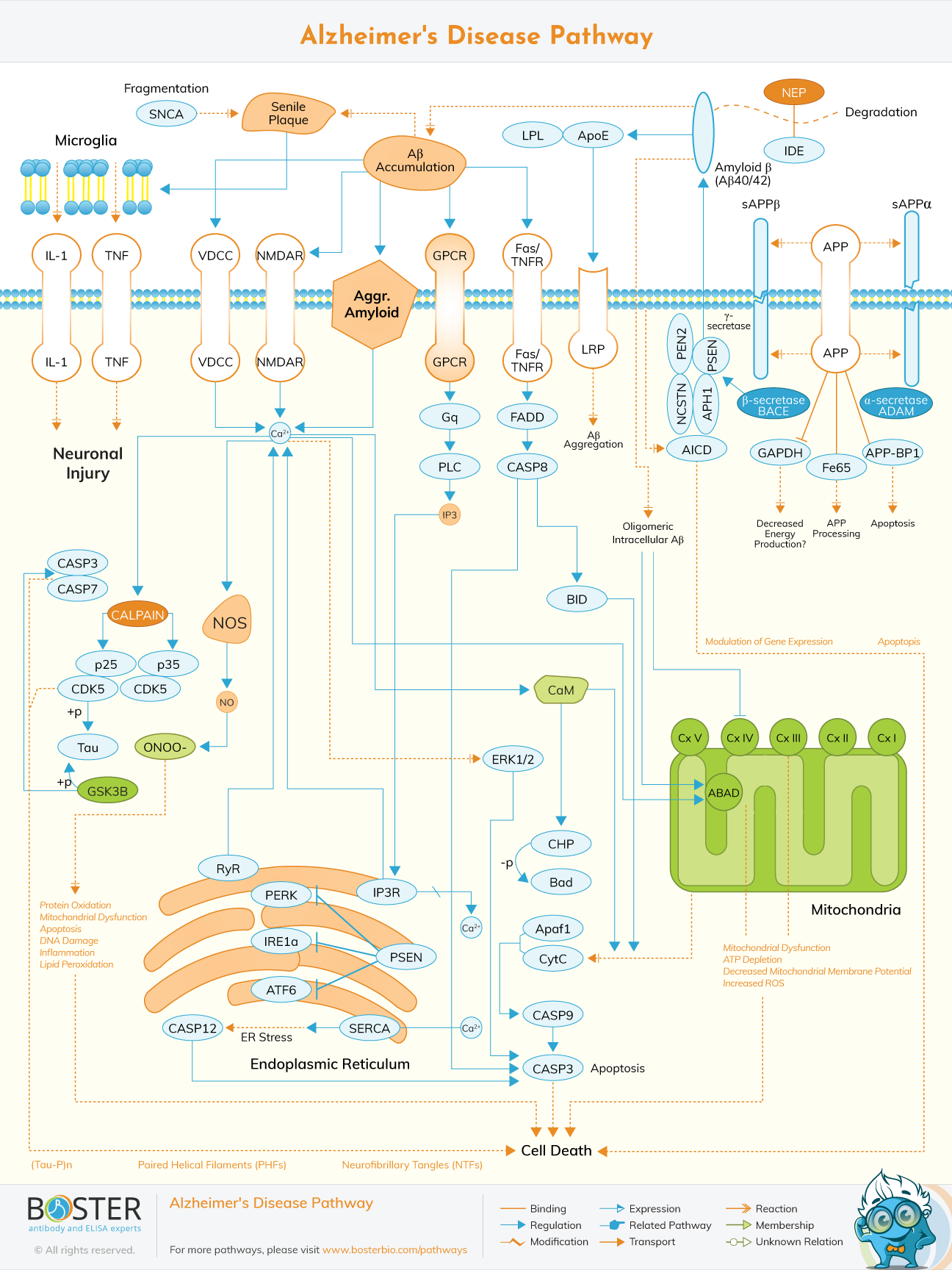
Kumar reviewed the pathophysiology and management of Alzheimer’s disease. In the paper, the development and promising therapies were discussed. Although there is enough information about the disorder, only limited choices exist for its management (Kumar, 2015). AChEls and Memantine, which are medications for its treatment, only treat symptoms and neglect the underlying factors responsible for the disease. However, the administration of vaccine seems positive because patients with early stages show improvement in their behaviour after Ab vaccination (Kumar, 2015). New solutions under verification include NOS modulation, and DNA vaccination (Kumar, 2015).
Also, Mizuno constructed an Alzheimer’s disease pathway named “AlzPathway” which is accessible by everyone. The pathway is a directory of Alzheimer’s disease signaling pathway. It is the alpha and omega for decoding pathogenic of Alzheimer’s disease (Satoshi, 2015). The pathway is made up of 1347 molecules, 1070 reactions and 129 phenotypes in neuron, brain blood barrier, astrocyte synaptic, microglial cells and their positions.
In 1901, Alois Alzheimer, a German psychiatrist, recognized the first fifty-year-old woman with Alzheimer’s disease. After five years, eleven more cases were described in medical journals (Alzheimer, 1907). Back then, old age was regarded as a risk factor of increasing dementia. Emil kraepelin was the first to explain it as a typical disease after debunking some information in the medical literature of Auguste D (Berrios, 1990). Alzheimer’s disease also called presenile dementia is a category of senile dementia described by Kraeplin in one of his books (Kraepelin, 2007).
In the 20th century, Alzheimer’s disease diagnosis was restricted to those within the susceptible age group (45 to 65 years old) showing signs of dementia. After 1977, the concept was modified when a seminar on Alzheimer’s disease concluded that similar symptoms exist in senile and presenile dementia. Hence, this brought about the diagnosis of Alzheimer’s disease irrespective of age (Boller, 1998).
Alzheimer’s disease (AD) is said to be the result of unusual accumulation of proteins in the brain tissues. Among them is amyloid which accumulates to build up plaques in the brain tissues. Another type of protein involved is TAU which accumulates to form tangles in the brain tissues. Though the root of this process is still a mystery, the fact remains that it exists several years before the symptoms become visible.
Dysfunction of the brain tissues is also accompanied by reduction neurotransmitters that transmit information in the brain. Subjects with Alzheimer’s disease have low levels of acetylcholine (a neurotransmitter) in their brain. Subsequently, various sections of the brain shrivel especially the part that is in charge of memories. In special cases of Alzheimer’s disease, there is dysfunction of the brain cells and early symptoms include loss of vision or speech impairment.
There are four stages of clinical classification of AD described below.
The symptoms of AD include the continuous decline in episodic memory and other cerebral skills, causing world cognitive deterioration, changes in character and conduct. There are early onset and late onset of Alzheimer’s disease. Late onset occurs in people above 65 years while early onset manifests in younger people. Dementia associated with AD in these subjects begins around 50 to 60 years. Maturity and poor educational background are the significant risk factors for late manifestation of Alzheimer’s disease. Others include the existence of the allele ε4 of the apolipoprotein E (APOE) gene, prolonged cardiovascular risk factors, past occurrence of brain shock accompanied by unconsciousness, inactivity, and poor cognitive demand. On the contrary, AD in younger subjects is commonly related to genetic mutations which are usually the Amyloid Precursor protein (APP) gene and Presenilin.
Age doesn’t cause any disparity in clinical findings in AD patients. Akin clinical findings are recorded in AD patients irrespective of their age. The distinctive features of Alzheimer’s disease include the existence of neuritic plaques and neurofibrillary tangles, gliosis, neuronal loss and dystrophic neuritis as well. Neuritic plaques are exogenous lesions primarily consisting of -β42 peptide (Aβ42). Neurofibrillary tangles are endogenous wounds majorly made up of hyperphosphorylated TAU protein.
Current medications for Alzheimer’s disease are tacrine, galantamine, rivastigmine and donepezil (acetylcholinesterase inhibitors), NMDA receptor antagonist and memantine, though with little improvements in cognitive issues. The use of Aducanumad was recently permitted in the US. It is the first medical treatment approved for the disease because it works on the basic pathophysiology of the disorder. Acetylcholinesterase inhibitors act by decreasing ACh (Acetylcholine) catabolism which will increase the amount of acetylcholine in the brain and fight its loss due to cholinergic neurons. Mild to moderate side effects of these medications noticed in about 20% of its users include nausea and vomiting associated with cholinergic overload. The side effects of the medications can be controlled by regulating the dosage. Other rare side effects are bradycardia, low appetite, weight loss and more secretion of gastric acid.
Similarly, EGb 761 (derived from Ginkgo biloba) is generally used for treatment of Alzheimer’s disease in Europe. It is active against symptoms of Alzheimer’s disease and vascular dementia. EGb 761 performs a vital function alone or in combination with other therapies to improve their efficiency. It protects the neurons, eliminates free radicals, boosts mitochondrial activities and regulates the level of serotonin and dopamine. Clinical studies for its effectiveness against mild and moderate dementia revealed significant reduction in cognitive problems, improved daily and mental activities. Nevertheless, EGb 761has not been proven to stop the development of Alzheimer’s disease to dementia.
Amyloid precursor protein (APP) is an integral polytopic protein that is among the major proteins in the central nervous system (CNS). It is also present in the epithelium and blood cells (peripheral tissues). On the other hand, amyloid precursor protein can be produced via two ways which are the amyloidogenic pathways and the non-amyloidogenic pathways. In the latter, the enzyme γ-secretase divides APP into sAPPα (soluble N-terminal fragment) and C83 (C-terminal fragment). The α-secretase acts on C83 to produce 3KDa (C3) (C-terminal fragment). Besides, β-secretase can divide Amyloid precursor protein into sAPPβ (smaller N-terminal fragment) and C99 (C-terminal fragment) that creates the full-length Aβ (β-amyloid peptides) due to further division by γ-secretase.
There are different Aβ peptide species. Aβ40 and Aβ42 are majorly present in the brain. Aβ species are secreted as monomers which further become dimmers, trimers, protofibrils and fibrils which later accumulate, spread out and mature into amyloid plaques. Aβ40 and Aβ42 are closely related but Aβ42 is more susceptible to accumulation and fibrilization because it is most deadly Aβ peptide and performs a key function in the pathogenesis of Alzheimer’s disease.
The Aβ oligomers are regarded as the most lethal form of the Aβ peptide. They relate with neurons and glial cells causing inflammatory cascaded, non-regulated calcium metabolism, oxidative stress, introduction of neuronal apoptosis and TAU phosphorylation. The combination of the aforementioned brings about a continuous beneficial response window where the release of Aβ peptides causes harmful effects to the neuronal cells. That adds to the malfunctioned APP metabolism and subsequent secretion of Aβ peptides. Amyloid-β fibrils accumulate in neuritic plaques chronologically listed: diffuse neuritic plaques, mature neuritic plaques, senile plaques and phantoms of senile plaques in severe Alzheimer’s disease. Also, the plaque build-up has a toxic effect on neuronal homeostasis causing neuronal malfunction and death.
In the physiological circumstances, the APP is preferably broken down by the non-amyloidogenic pathway and a balance exists between the secretion of Aβ peptides and their removal from the cerebrum. Presently, Apolipoprotein E (APOE) and the insulin degrading enzyme (IDE) are the two proteins majorly responsible for the removal of Aβ peptides from the brain. The precise means by which Aβ peptides are removed from the brain is not explicit till date. However, a recent theory states that these proteins attach to Aβ peptides, preventing their accumulation into oligomers and ultimately leading to their removal from the brain. In the pathological circumstances, there is a metabolic change beneficial to the amyloidogenic division of APP which combines with reduced elimination of Aβ to cause the aggregation of Aβ in the brain tissue.
The TAU protein is a microtubule related to protein existing in most tissues. It is majorly released in the central and peripheral nervous system. It is a key part of the neuronal cytoskeleton which enables it to work together with α- and β-globulin and make the microtubules become stable. In neurons, the microtubules are vital for the management of neuronal structure, synaptic plasticity, and axonal transport. Six TAU isoforms are present in humans. The key disparity among these isoforms is the differences in the N-terminal end of the protein and the presence of three or four domains attached to tubulin. The relationship between TAU and tubulin is a self-motivated process where TAU supports self polymerization and slows down rapid depolymerization of the tubulin. The procedure is controlled by the phosphorylation condition of TAU protein which is made up of about 79 phosphorylation spots at threonine and serine deposits. The equilibrium between phosphorylation and dephosphorylation of these epitopes supports conformational modifications that affect the way TAU protein relates with α- and β-tubulin and that calms the microtubules in the neurons. A lot of protein kinases and phosphatases are part of the control of TAU phosphorylation. Enzyme GSK3β (Glycogen synthase kinase 3β) is the central TAU-kinase in the neurons.
A decreased action of certain phosphatases was also recorded in the cerebral tissues of subjects suffering from Alzheimer’s disease such as PP1, PP2A and PP5. The main deposits of serine and threonine that are present in a hyperphosphorylated state in harmful TAU and PHFs is accompanied by proline, indicating that TAU kinases is among the group of proline-controlled enzymes such as cyclin-dependent kinases (CDC2, CDK5, TPKll), MAPK (mitogen-activated kinases), and GSK3β. These kinds of enzymes can phosphorylate TAU outside a living organism (in vitro) and was found in the cellular strata of cerebral tissue of patients suffering from Alzheimer’s disease. Other kinases, such as PKC, casein-kinases I and II, calcium/calmodulin-stimulated protein kinase II (CaMPK-II) and protein kinase A (PKA), that are not directed by proline were also found in neurofibrillary tangles and are crucial to the control of TAU phosphorylation, adjusting the activity of the previous enzymes.
At embryonic phase, neuronal TAU is primarily found in an agitated phosphorylated condition. This is because of the increased need for neuroplastic modifications in neurons and synapses at initial developmental phases of the central nervous system (CNS). However, in the full-fledged central nervous system, dephosphorylated TAU is needed to keep microtubules steady and manage structural and functional cytoskeletal homeostasis.
Self-motivated alterations in the phosphorylation condition of TAU crops up in pathways used for neuronal changes and synaptic plasticity. For clinical diagnosis, TAU can be unusually hyperphosphorylated which affects its ability to attach to tubulin, causing the damage of the microtubule configuration, axonal movement and synaptic metabolism. Such alterations ultimately lead to cytoskeleton breakdown, failure of cell viability and neuronal death.
NFT (neurofibrillary tangles) which are intra-neuronal wounds as described by Alois Alzheimer are majorly made up of paired helical filaments (PHFs) of hyperphosphorylated TAU. 25 unusual phosphorylation areas or more was explained in Alzheimer’s disease representing a sign of neuronal degeneration in this condition. The unusual phosphorylation of TAU in serine or threonine deposits close to the tubulin binding domain supports disaggregation of the TAU-tubulin complex and rearranges TAU into paired helical filaments to form neurofibrillary tangles (NFT). Owing to the significance of TAU in neuronal stability and homeostasis, its hyperphosphorylation brings about a flow of activities that eventually leads to neuronal death.
In spite of reliable facts promoting the major role of Aβ peptides and hyperphosphorylated TAU in the pathogenesis of Alzheimer’s disease, none provide acceptable records for the complete range of pathological mechanisms related to this disease. NFTs and Aβ plaques may be a self-sufficient phenomenon in Alzheimer’s disease. So, other processes that were introduced that engage the action of upstream enzymes that are active in the aforementioned phenomenon. GSK3β is a vital enzyme in the maintenance of cellular metabolism including TAU phosphorylation. Wnt signaling is another essential arm of neurobiology of Alzheimer’s disease. Since Wnt signaling causes the deactivation of GSK, inhibiting TAU phosphorylation in the epitopes dependent on GSK. Regarding Alzheimer’s disease, GSK3β was discovered in a hyperactive condition causing the hyperphosphorylation of TAU. Besides, GSK3β controls APP metabolism and supports amyloidogenic division (for excess secretion of Aβ), decreases neurogenesis and amplifies apoptosis.
Alzheimer’s disease (AD) is the usual contributory factor to dementia among the aged. In the global aged population, about 24.3 million people experience dementia. In 2020, the statistics rose to 42.3 million people and expected to become 81.1 million by 2040. There are different facts that confirm the view that the malfunction in TAU homeostasis is a major occurrence in Alzheimer’s disease, an incident also present in similar neurodegenerative conditions like frontotemporal dementia and multiple system atrophy. Neuropathological research revealed that the circulation of NFTs in the brain correlates with the medical advancement of dementia in Alzheimer’s disease. Furthermore, intra-neuronal hyperphosphorylated TAU exists in the brain of patients with dementia that is not severe, without β-amyloid pathology. Hence, TAU hyperphosphorylation can be the initial occurrence in the physiopathology of Alzheimer’s disease (AD), while other processes including over secretion of Aβ, oxidative stress and start of inflammatory cascade may have lesser effects on the whole malfunction in neuronal homeostasis.
Cardiovascular diseases such as diabetes, hypertension, and hypercholesterolemia are linked with increased risk of manifestation or rapid degeneration of Alzheimer’s disease. Other risk factors include smoking and hormone replacement therapy for menopausal women.