This website uses cookies to ensure you get the best experience on our website.
- Table of Contents

1 Citations 11 Q&As


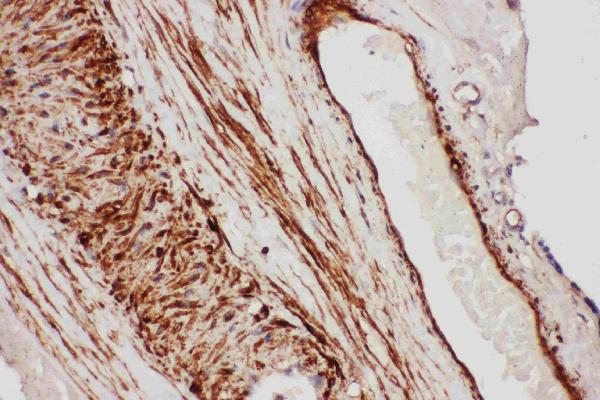
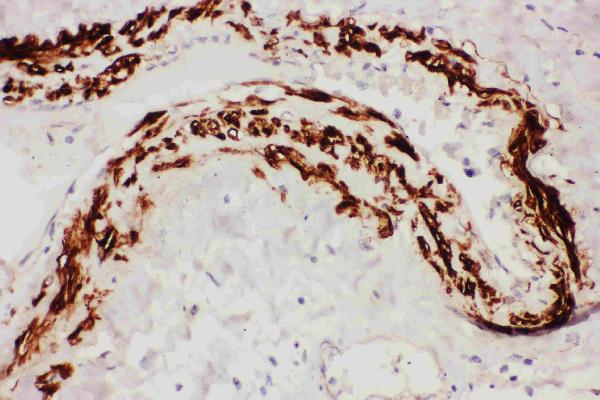
Facts about Tissue factor pathway inhibitor.

.
| Human | |
|---|---|
| Gene Name: | TFPI |
| Uniprot: | P10646 |
| Entrez: | 7035 |

| Belongs to: |
|---|
| No superfamily |

convertin; EPI; EPITFPI1LACITFI; Extrinsic pathway inhibitor; LACI; Lipoprotein-associated coagulation inhibitor; TFPI; tissue factor pathway inhibitor (lipoprotein-associated coagulation inhibitor); tissue factor pathway inhibitor
Mass (kDA):
35.015 kDA

| Human | |
|---|---|
| Location: | 2q32.1 |
| Sequence: | 2; NC_000002.12 (187464230..187554505, complement) |
Mostly in endothelial cells.
[Isoform Alpha]: Secreted.; [Isoform Beta]: Microsome membrane; Lipid-anchor, GPI-anchor.


PMID: 2452157 by Wun T.-C., et al. Cloning and characterization of a cDNA coding for the lipoprotein- associated coagulation inhibitor shows that it consists of three tandem Kunitz-type inhibitory domains.
PMID: 2781520 by Girard T.J., et al. Identification of the 1.4 kb and 4.0 kb messages for the lipoprotein associated coagulation inhibitor and expression of the encoded protein.
*More publications can be found for each product on its corresponding product page