This website uses cookies to ensure you get the best experience on our website.
- Table of Contents

Facts about Receptor-interacting serine/threonine-protein kinase 3.

RIPK3 binds to and enhances the action of three metabolic enzymes: GLUL, GLUD1, and PYGL. These metabolic enzymes can eventually stimulate the tricarboxylic acid cycle and oxidative phosphorylation, which could result in enhanced ROS production.
| Human | |
|---|---|
| Gene Name: | RIPK3 |
| Uniprot: | Q9Y572 |
| Entrez: | 11035 |

| Belongs to: |
|---|
| protein kinase superfamily |

EC 2.7.11.1; Receptor-interacting protein 3; receptor-interacting serine/threonine-protein kinase 3; receptor-interacting serine-threonine kinase 3; RIP3; RIP-3; RIP3receptor interacting protein 3; RIPK3; RIP-like protein kinase 3
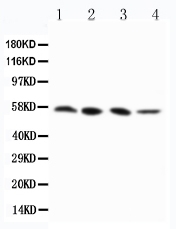
Mass (kDA):
56.887 kDA

| Human | |
|---|---|
| Location: | 14q12 |
| Sequence: | 14; NC_000014.9 (24336025..24339991, complement) |
Highly expressed in the pancreas. Detected at lower levels in heart, placenta, lung and kidney. Isoform 3 is significantly increased in colon and lung cancers.
Cytoplasm, cytosol. Cell membrane. Mitochondrion.



PMID: 10339433 by Yu P.W., et al. Identification of RIP3, a RIP-like kinase that activates apoptosis and NFkappaB.
PMID: 10358032 by Sun X., et al. RIP3, a novel apoptosis-inducing kinase.
*More publications can be found for each product on its corresponding product page