This website uses cookies to ensure you get the best experience on our website.
- Table of Contents




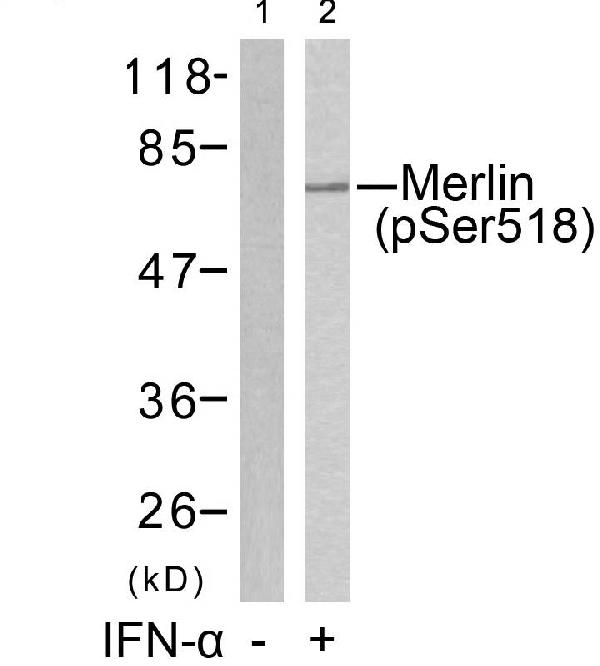
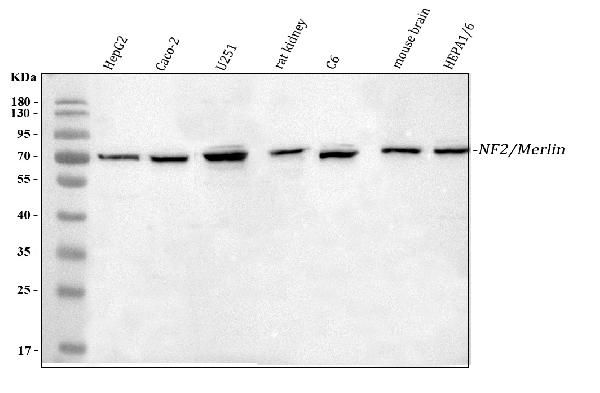
Facts about Merlin.

May act as a membrane stabilizing protein. May inhibit PI3 kinase by binding to AGAP2 and impairing its stimulating action.
| Human | |
|---|---|
| Gene Name: | NF2 |
| Uniprot: | P35240 |
| Entrez: | 4771 |

| Belongs to: |
|---|
| No superfamily |

ACN; BANF; Merlin; moesin-ezrin-radixin like; Moesin-ezrin-radixin-like protein; moesin-ezrin-radizin-like protein; neurofibromin 2 (bilateral acoustic neuroma); neurofibromin 2 (merlin); neurofibromin-2; NF2; SCH; Schwannomerlin; Schwannomin
Mass (kDA):
69.69 kDA

| Human | |
|---|---|
| Location: | 22q12.2 |
| Sequence: | 22; NC_000022.11 (29603556..29698600) |
Widely expressed. Isoform 1 and isoform 3 are predominant. Isoform 4, isoform 5 and isoform 6 are expressed moderately. Isoform 8 is found at low frequency. Isoform 7, isoform 9 and isoform 10 are not expressed in adult tissues, with the exception of adult retina expressing isoform 10. Isoform 9 is faintly expressed in fetal brain, heart, lung, skeletal muscle and spleen. Fetal thymus expresses isoforms 1, 7, 9 and 10 at similar levels.
[Isoform 1]: Cell projection, filopodium membrane; Peripheral membrane protein; Cytoplasmic side. Cell projection, ruffle membrane; Peripheral membrane protein; Cytoplasmic side. Nucleus. In a fibroblastic cell line, isoform 1 is found homogeneously distributed over the entire cell, with a particularly strong staining in ruffling membranes and filopodia. Colocalizes with MPP1 in non-myelin-forming Schwann cells. Binds with DCAF1 in the nucleus. The intramolecular association of the FERM domain with the C-terminal tail promotes nuclear accumulation. The unphosphorylated form accumulates predomi



PMID: 8453669 by Trofatter J.A., et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor.
PMID: 8379998 by Rouleau G.A., et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2.