This website uses cookies to ensure you get the best experience on our website.
- Table of Contents
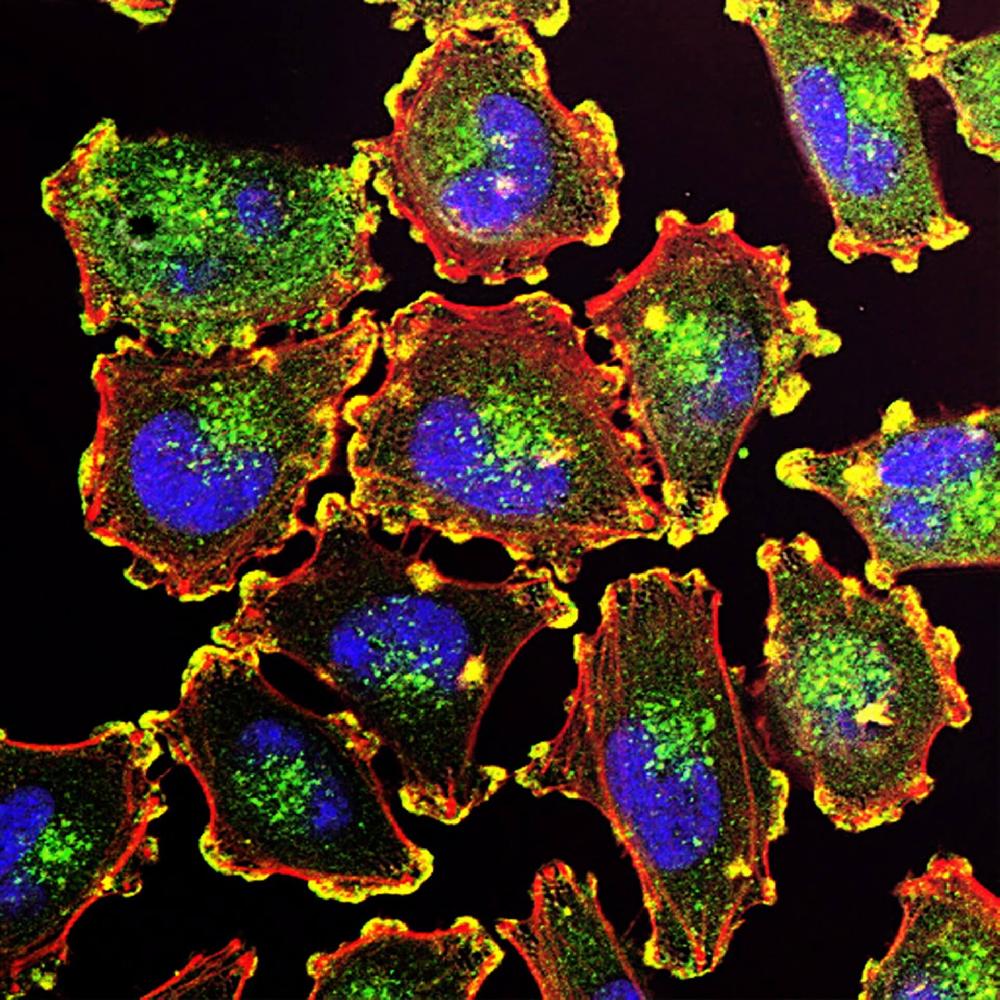

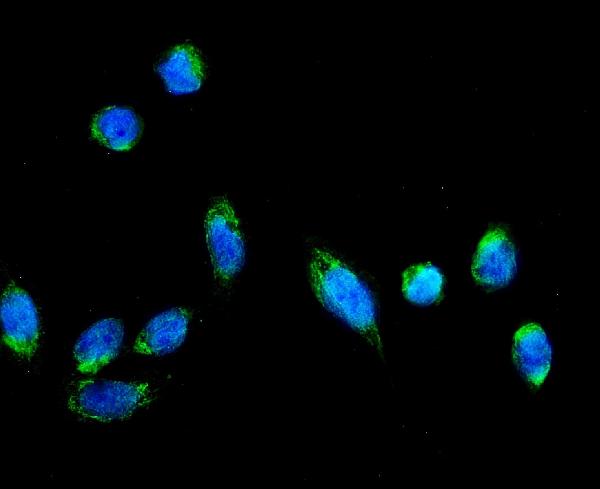
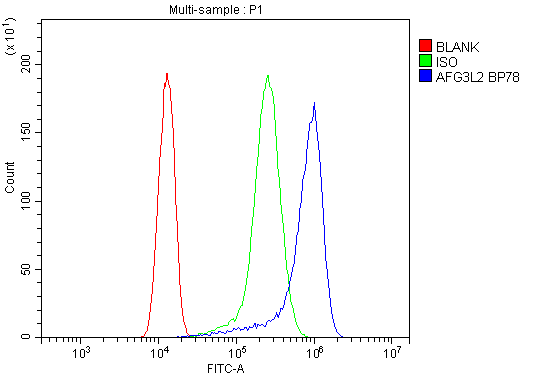

Facts about AFG3-like protein 2.

Required for the maturation of paraplegin (SPG7) after its cleavage by mitochondrial-processing peptidase (MPP), converting it into a proteolytically active mature form (By similarity). .
| Human | |
|---|---|
| Gene Name: | AFG3L2 |
| Uniprot: | Q9Y4W6 |
| Entrez: | 10939 |

| Belongs to: |
|---|
| No superfamily |

AFG3 (ATPase family gene 3, yeast)-like 2; AFG3 ATPase family gene 3-like 2 (S. cerevisiae); AFG3 ATPase family gene 3-like 2 (yeast); AFG3-like protein 2; ATPase family gene 3, yeast; EC 3.4.24; EC 3.4.24.-; FLJ25993; Paraplegin-like protein; SCA28; spinocerebellar ataxia 28
Mass (kDA):
88.584 kDA

| Human | |
|---|---|
| Location: | 18p11.21 |
| Sequence: | 18; NC_000018.10 (12328944..12377309, complement) |
Ubiquitous. Highly expressed in the cerebellar Purkinje cells.
Mitochondrion. Mitochondrion inner membrane; Multi-pass membrane protein.


Anti-AFG3-like protein 2 (AFG3L2) is an antiserum developed by Boster Bio. This antibody has been validated for IHC and IF applications. It has been shown to react with Human, Mouse, Rat, and rat samples. Using this antibody, you can perform quantitative immunohistochemistry analysis of the protein. In addition, Boster Bio's A04820 antibody has been used in research projects using antiserum to detect AFG3L2.
Boster Bio Anti-AFG3L2-Marker is part of a collection of primary antibodies for use in Immunohistochemistry, Western Blotting, and ELISA applications. This antibody has high affinity and is validated for use in multiple methods, including IF, ICC, and WB. For additional information, please see the product description. It is available in several formats.
The AFG3L2 gene has multiple functions. It is essential for the metabolism of energy and is involved in the production of glycogen, which is crucial for energy production in cells. The gene is expressed in a variety of cell types, including the developing brain, heart, liver, and kidney. Its mRNA is a common target for gene editing, but it is also an attractive option for studies of human disease.
AFG3L2 is a gene that encodes a protein. It is found in Drosophila melanogaster, Xenopus laevis, and Danio rerero. It is a paralog of the FtsH protein found in E. coli. The gene is found in all cells of all animal species. Its mutations are responsible for a range of human diseases.
The gene AFG3L2 is a mitochondrial protein. It is implicated in the processing of mitochondrially-coded respiratory chain subunits and in the assembly of ribosomes. The gene AFG3L2 is closely related to the paraplegin gene, which is responsible for a rare autosomal recessive form of hereditary spastic paraplegia. Several other diseases and disorders are linked to the AFG3L2 gene, including hereditary spastic paraplegia and hereditary spastic paraplegial disorders.
Antibodies for AFG3L2 have been developed to detect this protein. These antibodies are designed for use with Western blot analysis and protein transfer. They can detect a range of protein sizes. The antibodies are available in different strengths, so you can choose the one that works best for your studies. If you are considering purchasing this antibody, you should do so. It can be stored for up to a year or even six months.
The AFG3L2 gene is a candidate gene for the investigation of various neurodegenerative disorders. The AFG3L2 gene is related to many inherited retinal ganglion neuropathies, including glaucoma. Therefore, screening for AFG3L2 in patients with retinal degeneration is highly recommended. There are many applications of the AFG3L2 gene. If you are interested in using this gene, it will make your diagnostic process easier.
In recent research, Muona et al. identified a heterozygous c.1875G-A transversion in the AFG3L2 gene. This mutation alters the amino acid sequence of the C-terminal proteolytic peptidase-M41 domain. This change decreases the electrostatic potential difference between the matrix and inner mitochondrial membrane side of the hexameric complex, reducing its central pore dipole. The mutation was not found in 380 control chromosomes, but Muona and colleagues identified a cluster of patients with progressive myoclonic epilepsy.
AFG3L2 is a multigene marker. It is responsible for the regulation of the expression of several different proteins. The gene is present in Xenopus laevis, Drosophila melanogaster, and Danio rerero, which are known models of human disease. The mutation is also found in the bacterial protein FtsH. In humans, the gene SPG7 is a paralog of AFG3L2.
The Y616C mutant protein displayed impaired ability to assemble in a protease complex and lacked the ability to interact with paraplegin. The mutant protein's functional role in m-AAA protease assays was further extended. Moreover, Di Bella et al. discovered that AFG3L2 is highly expressed in the cerebellum of human, with expression in the deep cerebellar layer of the brain and pyramidal cortical neurons.
Another gene that is associated with optic neuropathies is SPG7. AFG3L2 and SPG7 are linked in most cases of dominant optic atrophy. In patients with this disease, exonic sequencing of the OPA1 gene leads to 50% positive results. More research is needed in this area to make an accurate genetic diagnosis of this condition. Moreover, m-AAA proteases might also be involved.
In this study, we used a polyclonal antibody against murine Afg3L2 and a Yta10 mitochondrial targeting sequence as a mAb. We found that the hAFG3L2 complex is capable of immunodetection of peptides. These results suggest that high-affinity antibodies against Afg3L2 and Yta10 are more specific than polyclonal antibodies.
To measure the amount of hAFG3L2 or AFG3L2E575Q in the eluate fraction, we used laser densitometry. The smaller hAFG3L2-containing complex was likely formed by partial dissociation of the larger complex. To calibrate the immunoblotting results, we used three marker proteins: Hsp60 (840 kDa), apoferritin (44 kDa), and bovine serum albumin (66 kDa). We also analyzed Ccp1 processing in yeast MrpL32 cells expressing hAFG3L2E575Q.
The two human m-AAA protease subunits hAFG3L2 and hAFG3L1 have been isolated from brain and liver mitochondria. The mitochondrial fractions were subjected to immunoblotting and SDS-PAGE. We also used preimmune antiserum as a negative control. The antibodies bind m-AAAA protease, a complex found in mitochondrial inner membrane that regulates protein quality and ribosome assembly. Paraplegin mutations lead to spastic paraplegia and axonal degeneration.
Mutations in AFG3L2 result in neurodegeneration, behavioral defects, and impaired mitochondrial transcription and translation. These phenotypes are partly rescued by overexpression of mito-UPR components and mitochondrial stress pathways. Consequently, high-affinity antibodies against AFG3L2 may be an important therapeutic tool in a range of human diseases resulting from mutated AFG3L2.
Afg3l1 and afg3l2 are quantitatively assembled with one another and may form a homo-oligomeric complex. However, the majority of Afg3l2 is outside the structure of Afg3l2 and is likely part of a hetero-oligomeric complex. The immunoreactivity of these antibodies was evaluated in a series of experiments using anti-O1-eFluor 660.
To demonstrate the activity of Afg3L2, we used a homo-oligomeric hAFG3L2 complex. This molecule forms a homo-oligomeric complex within the inner membrane of yeast cells. Using BN-PAGE, we determined the native molecular mass of hAFG3L2 and found that it was part of a complex with an approximate molecular mass of 900 kDa.
PMID: 10395799 by Banfi S., et al. Identification and characterization of AFG3L2, a novel paraplegin- related gene.
PMID: 14623864 by Atorino L., et al. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia.

